
Abschlussbericht zum Forschungsprojekt: Epidemiologie für Diphtherie-Erreger und Erreger der Lyme Krankheit
Projektbezeichnungen: Epidemiologie für Diphtherie-Erreger und Erreger der Lyme Krankheit
Projektlaufzeit: 01.10.2012-31.12.2013
Zusammenfassung
Zielsetzung des Projektes war die Etablierung von Multilocus Sequence Analysis (MLSA) für Corynebacterium diphtheriae, C. ulcerans, C. pseudotuberculosis und Borrelia burgdorferi sensu lato als zukunftsweisende Maßnahme für molekulare Epidemiologie und als Basis zur Verbesserung diagnostischer Methoden. Weiterhin sollten die Daten zur Beurteilung der Stammsammlungen des Konsiliarlabors Diphtherie und des NRZ für Borrelien in einem globalen Kontext dienen.
Multilocus Sequence Analysis (MLSA) für Corynebakterium
Das für C. diphtheriae existierende MLSA System, das in der PubMLST.org Datenbank vorhanden ist, wurde daher übernommen. Das MLSA System beruht auf der Sequenzanalyse von sieben konservierten Genen, nämlich atpA, dnaE, fusA, odhA, rpoB, dnaK und leuA.
C. diphtheriae Stämme (n=65) der Stammsammlung wurden mittels bestehender Primer amplifiziert und sequenziert. Sequenzauswertung erfolgt im Rahmen einer Doktorarbeit (Christina König).
Für C. pseudotuberculosis konnte für 11 Stämme eine Amplifizierung erfolgreich durchgeführt werden. Die Daten werden im Rahmen der Doktorarbeit von Frau König ausgewertet.
Analyse von C. ulcerans. Um eine möglichst hohe Vergleichbarkeit zwischen Corynebakterienspezies zu erhalten, wurde bei der Entwicklung und Etablierung eines MLSA Systems für C. ulcerans und C. pseudotuberculosis darauf geachtet, die gleichen Gene wie für C. diphtheriae zu verwenden. Für fünf der sieben konservierten Gene konnten die Primer von C. diphtheriae verwendet werden. Primer für C. ulcerans für die beiden Gene dnak und leuA wurden speziell anhand der C. ulcerans Vollgenomsequenz des Stammes 809 aus GenBank designed und die MLST PCR für diese Spezies am LGL etabliert. Insgesamt wurden bei 44 Isolaten der Stammsammlung des Konsilliarlabors Diphtherie (NCLoD), (n=31 von Humanpatienten und n=13 von Tieren) mittels PCR erfolgreich die MLST Gene amplifiziert und sequenziert.
Die Sequenzen wurden in die MLST Datenbank (http://pubmlst.org/cdiphtheriae/) eingereicht, um Allelenummern und Sequenztypnummer zu erhalten. Die Sequenzen wurden mit der Software Phyloviz analysiert, um Korrelationen von Diphtherie-Toxinpositivität (Abb. 1A) und Sequenztyp sowie biologische Herkunft (Human, tierisch; Abb. 1B) zu visualisieren.
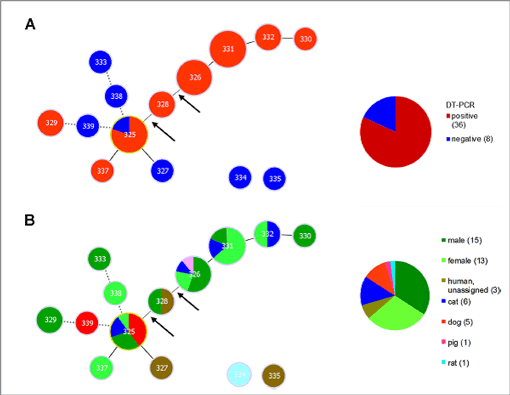
Abbildung 1 zeigt Diagramme der MLST Daten von C. ulcerans Isolaten hergestellt in Phyloviz. (A) Das Diagramm verdeutlicht die Korrelation von Sequenztypen mit Diphtherietoxin (DT)-Positivität or –Negativität. Nur ein Sequenztyp (ST325) besteht aus sowohl DT-positive und –negative Stämmen. (B) der gleiche Sequenztyp (ST325) zeigt ein breites Spektrum an Wirten. Die meisten anderen ST, von denen mehr als ein Isolat vorliegt, wurden sowohl von Menschen als auch Tieren isoliert. Die Daten deuten nicht auf eine Korrelation zwischen Wirtsspektrum und Toxizität hin.
Ein phylogenetischer Baum, der von den zusammengefassten Sequenzdaten der sieben Gene angefertigt wurde, verdeutlicht die enge Verwandtschaft der DT-positiven Stämme. Die Daten wurden in einer Publikation zusammengefasst (König et al. 2014).
Das für C. ulcerans entwickelte MLST System wurde in einer Kooperationsarbeit zusammen mit der Arbeitsgruppe von Herrn Eisenberg zur Typisierung von Stämmen herangezogen, die aus einer Kolonie von Wasserratten isoliert worden waren. Die Ergebnisse wurden in BMC Microbiology publiziert (Eisenberg et al. 2015).
Wesentliche Erkenntnisse
- MLSA ist eine geeignete Methode, um Einsichten in die Verwandtschaftsverhältnisse bakterieller Erreger zu bekommen.
- Durch MLSA konnte gezeigt werden, dass DT-positive C. ulcerans Stämme eng verwandt sind, was die Vermutung zulässt, dass das toxin-codierende Gene, wenn es einmal erworben wurde, an Nachkommen weitervererbt wird.
- DT-positive Stämme, die denselben Sequenztyp darstellen, werden in einer Anzahl unterschiedlicher Wirte gefunden
MLSA für den Borrelia burgdorferi sensu lato Spezieskomplex
Für den Komplex der Lyme borreliose Spirochäten wurde ein MLSA System etabliert, das ebenfalls in der PubMLST.org Datenbank vorhanden ist (http:// pubmlst.org/borrelia/) und von einer NRZ Mitarbeiterin (Dr. Margos) betreut wird.
Die Methode (PCR Amplifizierung, Sequenzierung und Sequenzanalyse mittels entsprechender Software) wurde erfolgreich im Nationalen Referenzzentrum für Borrelien am LGL in Oberschleissheim etabliert. Insgesamt wurden 74 B. garinii (n=44) und B. bavariensis (n=30) Stämme sowie 138 Stämme von B. burgdorferi sensu stricto (Patientenisolate n=35, Zecken-DNA n=25) und B. afzelii (Patientenisolate n=78) mittels MLST typisiert.
Die Auswertung der B. bavariensis Isolate bestätigte die genetische Homogenität der Europäischen Population von B. bavariensis und diente als Grundlage zur Validierung der Spezies (Margos et al. 2013). Diese Arbeit wurde in Kooperation mit Kollegen aus dem Beijing Institute of Microbiology and Epidemiology und dem Beijing Institute of Disease Control and Prevention publiziert (Margos et al. 2013).
Die Auswertung der B. afzelii und B. burgdorferi sensu stricto Sequenzen ergab, dass Europäische B. burgdorferi sensu stricto Stämme im Vergleich zu B. afzelii signifikant häufiger an Symptomatik beteiligt sind, die auf Invasivität der Borrelien schliessen lassen. Die Anwendung von MLSA auf Europäische Borrelien Stämme erlaubte eine vergleichende Analyse mit Stämmen aus USA, die sich bereits in der Datenbank befanden. Dabei wurde festgestellt, dass von einigen Patienten Stämme isoliert wurden, die eng verwandt mit amerikanischen Stämmen sind. Weitergehende Untersuchungen mittels Vollgenomsequenzierung könnten Aufschluss darüber geben, ob diese Patienten die Infektion evtl. im Ausland erworben haben (siehe Report Typisierung von Neuroborrelioseerregern; Jungnick et al. 2015).
Die Verwandtschaft von europäischen und amerikanischen B. burgdorferi s.s. Stämmen ist aus der folgenden Abbildung (Abb. 2) ersichtlich, die auch verdeutlicht, dass einige Patientenisolate aus Europa denselben Sequenztyp aufweisen, wie amerikanische Stämme.

Abbildung 2: Die Abbildung zeigt einen Überblick über die Verwandtschaft von europäischen und amerikanischen B. burgdorferi s.s. Isolaten von Patienten und Zecken. Die Pie-chart links zeigt den Anteil der Proben unterschiedlicher Herkunft. In ST1 und ST3 ist ein Anteil EU HI festzustellen.
Farbkodierung:  USA Zecken (n= 456);
USA Zecken (n= 456);  USA Humanisolate (n=146);
USA Humanisolate (n=146);  Europa Zecken (n= 45);
Europa Zecken (n= 45);  Europa Humanisolate (HI) (n=33)
Europa Humanisolate (HI) (n=33)
Die B. garinii und B. bavariensis Daten werden derzeit im Rahmen einer Doktorarbeit weiter analysiert (M. Rieger).
Weiterführende Aktivitäten, die aufgrund des Projektes entstanden sind
Im Rahmen dieses Projektes wurden drei Doktorarbeiten (Sabrina Jungnick, Melissa Rieger, Christina König) vergeben und werden derzeit bearbeitet. Zwei Bachelorarbeiten (E. Dzaferovic – Eignung molekularer Marker zur Differenzierung von Borrelia Spezies; Isabell Notter – Entwicklung eine real-time PCR basierend auf MLST Genen zur Speziesidentifizierung von Borrelia) wurden ebenfalls vergeben, von denen eine bereits erfolgreich abgeschlossen wurde (Dzaferovic).
Durch die Etablierung von MLSA für Borrelia im Rahmen des Projektes konnten einige Genospecies validiert werden (z.B. Borrelia kurtenbachii sp. nov., Margos et al. 2014; Borrelia yangtzensis sp. nov., Margos et al. 2015). In Zusammenarbeit mit Prof. R.S. Lane, University of California Berkeley, und Prof. J. Piesman, CDC Fort Collins, konnten wir B. bissettii und B. californiensis als neue Spezies validieren (Publikation beim Internationalen Journal for Systematic and Evolutionary Microbiology eingereicht). Darüberhinaus wurde MLSA für Rückfallfieberborrelien, z.B. Borrelia persica in Kooperation mit Prof. Straubinger von der LMU (Schwarzer et al. 2015) oder Borrelia miyamotoi, entwickelt und etabliert werden. Derzeit wird MLSA für die Diagnostik von B. recurrentis (Läuserückfallfieber) eingesetzt.
Wesentliche Erkenntnisse
- B. burgdorferi sensu stricto wird häufiger als B. afzelii bei invasiver Borreliosesymptomatik gefunden, insbesondere Fälle von Neuroborreliose sind häufiger in Europa als in USA
- Einige Stämme, die von Patienten in Europe isoliert wurden, zeigen eine hohe genetische Ähnlichkeit mit Stämmen aus USA. Dies hat zu der Vermutung geführt, dass evtl. einige Patienten ihre Infektion in Nordamerika erworben haben.
- B. bavariensis besteht aus zwei Populationen, einer heterogenen asiatischen Population und einer genetisch homogenen Europäischen Population. Dies ist vermutlich auf Adaptation an einen neuen Vektor (Ixodes ricinus) zurückzuführen, gefolgt von geografischer Ausbreitung in dessen Verbreitungsgebiet (Europa). Einfluss auf Pathogenität wird in weiterführenden Projekten untersucht.
- Der B. burgdorferi sensu lato Spezieskomplex ist genetisch und ökologisch sehr heterogen, die Auswirkungen dieser Heterogenität auf Humanpathogenität sollten weiter untersucht werden.
Verknüpfung mit anderen Projekten
Das Projekt "Typisierung von Neuroborrelioseerregern" profitierte unmittelbar von den Auswertungen des oben beschriebenen Projektes, da diese Daten in die Entscheidung, welche Stämme mit Vollgenomsequenzierung (next generation sequencing; NGS) analysiert werden sollten. Aufgrund dieses Projektes werden demnächst zwei Bachelorarbeiten (Christoph Mang, Sabrina Hepner) am LGL betreut werden. Eine Kooperation besteht mit Dr. Nicholas Ogden, Public Health Canada, dessen Arbeitsgruppe die Invasion von Borrelia burgdorferi in Canada mittels MLSA untersucht. Es wird vermutet, dass Klimawandel eine Rolle bei der Ausbreitung von Vektor und Pathogen spielt.
Durch die Erkenntnisse, die im Rahmen der beiden Projekte (Molekulare Typisierung von Diphtherie-Erregern und Erregern der Lyme Borreliose, Typisierung von Neuroborreliose-Erregern) konnten Mittel von außerhalb eingeworben werden:
• Ein beim DAAD eingereichter Antrag, der die "Molecular Epidemiology of Lyme Borreliose spirochetes in Europe" zum Thema hat wurde positiv beschieden (Euro 10.000). Das Projekt wird in Kooperation mit der Slovak Academy of Sciences, Bratislava, Slovakia, durchgeführt.
• Ein ESCMID SG Antrag mit dem Titel "Exploring the genomics tool box for tick-borne bacterial pathogens of the Borrelia burgdorferi sensu lato species complex” wird mit Euro 30,000 gefördert.
Publikationen
Im Rahmen des Projektes wurden 8 Publikationen in englisch-sprachigen Fachzeitschriften mit Verweis auf die Förderung durch das StMGP angenommen.
Im Rahmen der Förderung publizierte Fachartikel in Journals mit Peer Review:
Christina König, Dominik M. Meinel, Gabriele Margos, Regina Konrad, Andreas Sing. 2014. Multilocus Sequence Typing of Corynebacterium ulcerans Provides Evidence for Zoonotic Transmission and for Increased Prevalence of Certain Sequence Types among Toxigenic Strains. Journal of Clinical Microbiology 52: 4318-4324
Tobias Eisenberg, Norman Mauder, Matthias Contzen, Jörg Rau, Christa Ewers, Karen Schlez, Gisa Althoff, Nicole Schauerte, Christina Geiger, Gabriele Margos, Regina Konrad, Andreas Sing. 2015. Outbreak with clonally related isolates of Corynebacterium ulcerans in a group of water rats. BMC Microbiology 15: 42
Gabriele Margos, Bettina Wilske, Andreas Sing, Cecilia Hizo-Teufel, Wu-Chun Cao, Chenyi Chu, Holger Scholz, Reinhard K. Straubinger, Volker Fingerle. 2013. Borrelia bavariensis sp. nov. is widely distributed in Europe and Asia. International Journal of Systematic and Evolutionary Microbiology 63, 4284–4288
Gabriele Margos, Joseph Piesman, Robert S. Lane, Nicholas H. Ogden, Andreas Sing, Reinhard K. Straubinger, Volker Fingerle. 2014. Borrelia kurtenbachii sp. nov.: A widely distributed member of the Borrelia burgdorferi sensu lato species complex in North America. International Journal of Systematic and Evolutionary Microbiology 64, 128–130
Gabriele Margos; Katrin Binder; Eldina Dzaferovic; Cecilia Hizo-Teufel; Andreas Sing; Manfred Wildner; Volker Fingerle; Keith A Jolly. 2015. PubMLST.org - the new home for the Borrelia MLSA database. Ticks- and Tick-borne Diseases 6, 869–871
Sandra Schwarzer, Gabriele Margos, Evelyn Overzier, Volker Fingerle, Gad Baneth, Reinhard K. Straubinger. 2015. Borrelia persica: In vitro cultivation and characterization via conventional PCR and multilocus sequence analysis of two strains isolated from a cat and ticks from Israel. Ticks and Tick-borne Diseases 6, 751-757
Regina Konrad, Stefan Hörmansdorfer, Andreas Sing. 2015. Possible human-to-human transmission of toxigenic Corynebacterium ulcerans. Clin Microbiol Infect. 21: 768-771
Sabrina Jungnick, Gabriele Margos, Melissa Rieger, Eldina Dzaferovic, Stephen Bent, Evelyn Overzier, Cornelia Silaghi, Georg Walder, Franziska Wex, Johannes Koloczec, Andreas Sing, Volker Fingerle. 2015. Borrelia burgdorferi sensu stricto and Borrelia afzelii: Population structure and Differential pathogenicity. International Journal of Medical Microbiology (in press)