
Aufklärung von Listeriose-
Ausbruchsgeschehen mittels
Next-Generation-Sequencing
Die am LGL bereits seit mehreren Jahren etablierte Next-Generation-Sequencing (NGS)-Technologie ermöglicht die Sequenzierung ganzer Bakteriengenome
und schafft damit die Voraussetzung für eine hochauflösende
Typisierung von Bakterienisolaten. Bei der NGS-basierten Typisierung können verschiedene Genombereiche mit einbezogen werden.
Ein möglicher
Ansatz ist beispielsweise die cgMLST-Analyse (Core Genome Multilocus Sequence Typing). Diese Analyse bezieht sich auf das Kerngenom der untersuchten Bakterienspezies, welches die Gene enthält, die in möglichst allen Isolaten einer Spezies zu finden sind.
Die DNA-Sequenz jedes dieser Gene kann zwischen verschiedenen Isolaten variieren. Auf Basis dieser Sequenzvarianten von Genen (Allele) können Bakterienisolate
in unterschiedliche Typen eingeordnet werden. Der Vergleich der Allele des Kerngenoms
verschiedener Bakterienisolate wird am LGL vor allem
zur Unterstützung von Ausbruchsanalysen genutzt.
Sehr ähnliche DNA-Sequenzen sind dabei ein Zeichen für eine nahe Verwandtschaft von Isolaten. Weisen zum Beispiel klinische und von Lebensmitteln gewonnene Listerien-Isolate ein nahezu identisches Allelprofil auf, kann dies einen Hinweis auf die potenzielle Infektionsquelle geben. Auf Basis der mittels NGS generierten Genomsequenz können noch weitere vergleichende Analysen vorgenommen werden, die zusätzliche Bereiche des Bakteriengenoms berücksichtigen. Auch hier sollten Isolate eines Ausbruchsgeschehens möglichst wenige Unterschiede aufweisen. Der Zusammenhang zwischen Lebensmittel und Infektionsquelle muss abschließend durch den Abgleich mit den Ergebnissen der epidemiologischen Untersuchungen (zum Beispiel Nennung des Lebensmittels bei Patientenbefragungen) geklärt werden.
In Zusammenarbeit mit dem Bundesinstitut ür Risikobewertung und dem Robert Koch-Institut konnte 2020 so unter anderem die Infektionsquelle eines internationalen Listeriose-Ausbruchsgeschehens identifiziert werden, wodurch es zu einem Rückruf des entsprechenden Produktes kam.
Die Ergebnisse der cgMLST-Analyse zweier Patientenisolate eines Ausbruchsgeschehens, des als ursächlich identifizierten Lebensmittels sowie weiterer Lebensmittel-Isolate sind in der Abbildung grafisch dargestellt. Die Zahlen geben an, um wie viele Allele sich die jeweils verbundenen Isolate unterscheiden.
Die Isolate des Ausbruchsgeschehens sind grau hinterlegt.
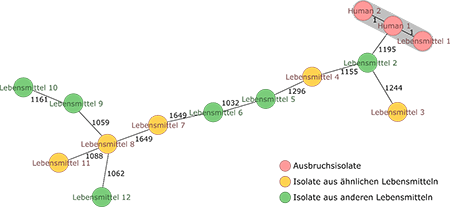
Abbildung 1: Darstellung der cgMLST-Analyse von Ausbruchsisolaten und Lebens-mittelisolaten ohne Zusammenhang mit dem Ausbruchs¬geschehen
Das ursächliche Lebensmittelisolat unterscheidet sich bei der Untersuchung von 1.701 Genen um maximal zwei Allele von den Patientenisolaten. Andere, zur Veranschaulichung willkürlich ausgewählte Isolate aus ähnlichen oder unterschiedlichen Lebensmittelprodukten unterscheiden sich deutlich von den Isolaten des Ausbruchsgeschehens.