
Fünf Jahre molekulare Epidemiologie mittels NGS und MLST am LGL
Hintergrund
Das European Centre for Disease Prevention and Control (ECDC) fordert als zukunftsweisendes Konzept, molekulare Techniken als Werkzeuge bei der epidemiologischen Untersuchung und Überwachung von Infektionskrankheiten zu integrieren. Die molekulare Charakterisierung und Feintypisierung von bakteriellen Krankheitserregern sind eine entscheidende Voraussetzung dafür. Deshalb haben das Nationale Referenzzentrum (NRZ) für Borrelien und das Konsiliarlabor für Diphtherie am LGL bereits 2012 erfolgreiche Projekte beim Bayerischen Staatsministerium für Gesundheit und Pflege beantragt, um solche neuartigen, molekularbiologischen Technologien zu erproben und zu etablieren. 2013 wurden dann die notwendigen Geräte beschafft.
NGS und MLST – Die Methoden:
Next Generation Sequencing (NGS) ist eine molekularbiologische High-Tech-Methode. Das Erbgut ganzer Organismen kann damit in Hochdurchsatz – also sehr schnell und in höchstmöglicher Auflösung untersucht werden. Das LGL hat in seinen PHM (Public Health Mikrobiologie)-Laboratorien diese zukunftsweisende Technologie, für deren komplexe Datenverarbeitung neben hochmodernen Laborgeräten immense Datenverarbeitungskapazitäten und Spezialexpertise in Bioinformatik nötig sind, seit 2013 als erstes Landesamt in Deutschland implementiert und konsequent ausgebaut. Es gehört damit zu den führenden PHM-Laboratorien in Europa und ist in Sachen NGS im Bereich des Öffentlichen Gesundheitsdienstes (ÖGD), Infektionserreger und PHM deutschlandweit Hauptansprechpartner des Robert-Koch Instituts..
Beim Multilocus Sequence Typing (MLST) werden mehrere (meist sieben oder acht) relativ stabile Gene (sog. house-keeping genes) im Genom bakterieller Erreger für epidemiologische Untersuchungen analysiert. Diese Gene werden in ihrer Basenabfolge sequenziert und Mutationen (Erbgutveränderungen) durch Vergleich mit elektronisch verfügbaren Gensequenzen von entsprechenden Bakterienstämmen ermittelt, die in internationalen Datenbanken hinterlegt und aufwändig gepflegt werden (z.B. https://www.pubmlst.org). Die Analyse solcher Mutationen (Einzelbasenunterschiede) erlaubt Aussagen zur verwandtschaftlichen Beziehung der untersuchten Bakterienstämme. Die internationale Borrelia MLST Datenbank an der Universität Oxford ( https://www.pubmlst.org/borrelia) wird von Dr. Gabriele Margos (
Anwendung von NGS oder MLST zur Ausbruchsuntersuchung und Identifizierung möglicher Infektionsquellen
Mit den hochauflösenden genomischen Daten können bei Ausbruchsuntersuchungen frühzeitig Hypothesen über mögliche Infektionsquellen aufgestellt werden, die dann durch epidemiologische Untersuchungen bestätigt werden müssen. Dies ist insbesondere bei zeitlich oder geografisch weit verstreuten Einzelerkrankungen wichtig, bei denen keine eingrenzbare Exposition zu einer gemeinsamen Infektionsquelle (z.B. Verzehr von Speisen bei einer Familienfeier) vorliegt. Konkret werden dabei für einen möglichen Ausbruch verantwortliche Bakterien-Isolate aus verschiedenen Materialien (z.B. humane Proben wie Stuhl, tierische Proben und Lebensmittelproben) sequenziert und ihre Genomsequenzen miteinander und in Referenz zu einer ausreichend großen Zahl anderer Stämme der gleichen Art verglichen. Je ähnlicher die Sequenzen, desto näher verwandt sind die Stämme. Die Ergebnisse werden üblicherweise in phylogenetischen Bäumen veranschaulicht (s. Abb.). Dies erlaubt Aussagen darüber, ob Erregerisolate von Patienten, Tieren oder Lebensmitteln genetisch miteinander zusammenhängen und so mit Unterstützung epidemiologischer Daten auf eine gemeinsame Infektionsquelle zurückgeführt werden können.
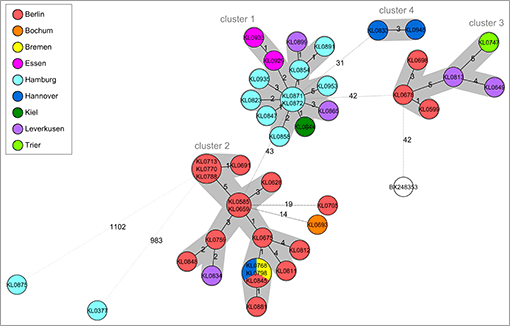
Abb.: Phylogentischer Stammbaum (Minimum Spanning Tree) zur Aufklärung eines überregionalen Ausbruchs mit Corynebacterium diphtheriae (Dangel, Berger et al., 2018, EID, im Druck). Die Alleldistanzen zwischen den als Kreise dargestellten Proben sind abgebildet und Ausbruchscluster mit Alleldifferenzen ≤ 5 grau hinterlegt. Die Farbcodierung kennzeichnet die Stadt der Probeneinsendung. Anhand des Baumes konnten vier Ausbruchscluster identifiziert werden innerhalb derer Erregerübertragung stattgefunden hat. Die Cluster beinhalten hauptsächlich Proben erkrankter Patienten aus Hamburg (Cluster 1) oder Berlin (Cluster2 und 3), aber auch weiteren norddeutschen Städten. Jede Probe mit eigenständigem genetischem Allelmuster ist als Kreis dargestellt. Proben mit identischem Allelmuster bilden einen gemeinsamen Kreis. Die Alleldistanzen zwischen Proben sind abgebildet und Ausbruchscluster mit Alleldifferenzen ≤ 5 grau hinterlegt. Die Stadt der Probeneinsendung ist farblich markiert. Der Baum zeigt vier Ausbruchscluster, innerhalb derer Übertragung eines Erregerstammes stattgefunden hat. Cluster 1 beinhaltet hauptsächlich Proben von erkrankten Patienten aus Hamburg (n=9), aber auch einzelne Proben aus Kiel (n=1), Leverkusen (n=2) und Essen (n=2). Cluster 2 und 3 beinhalten hauptsächlich Proben mit Berliner Herkunft (n=15 bzw. 3), sowie Beteiligung von Bremen, Hannover, Leverkusen und Bochum (n= je 1) in Cluster 2 sowie aus Leverkusen (n=2) und Trier (n=1) in Cluster 3. Cluster 4 besteht aus zwei Proben aus Hannover.
Aufgeklärte Krankheitsausbrüche
Seit 2014 konnten das PHM-Labor am LGL und das Konsiliarlabor für Diphtherie mehrere Ausbrüche mittels NGS aufklären:
- Ein großes Ausbruchsgeschehen in Norddeutschland, dessen Ursache Diphtherietoxin-negative Corynebacterium diphtheriae waren. Bei diesem Ausbruch traten teilweise schweren Wundinfektionen und Septitiden („Blutvergiftungen“) (14) auf.
- Zwei lebensmittelübertragene Salmonellose-Ausbrüche in bayerischen Gemeinschaftseinrichtungen. Das LGL identifizierte die ursächlichen Lebensmittel.
- Ein binationales Ausbruchsgeschehen mit Corynebacterium diphtheriae bei Asylbewerbern aus der Schweiz und aus Deutschland (9).
- Zoonotische Infektionsketten von Corynebacterium ulcerans (2, 8);
- Influenza (Grippe)-Ausbrüche in stationären Einrichtungen in Bayern (13).
Forschungsergebnisse
Am LGL führen das
Dies alles zeigt, dass die in den PHM-Laboratorien des LGL neu etablierten molekularen High-Tech-Typisierungsverfahren mit ihren hochauflösenden Daten dem Öffentlichen Gesundheitsdienst, dem Infektionsschutz, der Bevölkerung und der Forschung dienen.
Der Bereich molekulare Typisierungsverfahren wird kontinuierlich weiterentwickelt und neue Technologien etabliert, die die Sequenzierung noch größerer Genomabschnitte erlauben, als mit den aktuellen Methoden bisher erreichbar sind erlauben. Dadurch soll in Zukunft auch die Beschreibung von Gesamtgenomen selbst bislang unbekannter Bakterien ermöglicht werden.
Ausgewählte Publikationen der PHM Labore des LGL:
- Bolt F, Cassiday P, Tondella ML, Dezoysa A, Efstratiou A, Sing A, Zasada A, Bernard K, Guiso N, Badell E, Rosso ML, Baldwin A, Dowson C. 2010. Multilocus sequence typing identifies evidence for recombination and two distinct lineages of Corynebacterium diphtheriae. J Clin Microbiol.;48(11):4177-4185
- Meinel DM, Konrad R, Berger A, König C, Schmidt-Wieland T, Hogardt M, Bischoff H, Ackermann N, Hörmansdorfer S, Krebs S, Blum H, Margos G, Sing A. 2014. Zoonotic transmission of a toxigenic Corynebacterium ulcerans strain identified by Next Generation Sequencing. Emerg Infect Dis 21(2):356-358
- König C, Meinel DM, Margos G, Konrad R, Sing A. 2014. Multilocus sequence typing of Corynebacterium ulcerans provides evidence for zoonotic transmission and for increased prevalence of certain sequence types among toxigenic strains. J Clin Microbiol.;52(12):4318-4324
- Gatzmann F, Metzler D, Krebs S, Blum H, Sing A, Takano A, Kawabata H, Fingerle V, Margos G, Becker NS. 2015. NGS population genetics analyses reveal clonal evolution of a Lyme Borreliosis agent in Europe. Ticks Tick Born Dis 6 (3): 344-351
- Castillo-Ramírez S, Fingerle V, Jungnick S, Straubinger RK, Krebs S, Blum H, Meinel D, Hofmann H, Gürtler P, Sing A, Margos G. 2015. Trans-Atlantic exchanges have shaped the population structure of the Lyme disease agent Borrelia burgdorferi sensu stricto. Sci Rep 6:22794
- Margos G, Binder K, Dzaferovic E, Hizo-Teufel C, Sing A, Wildner M, Fingerle V, Jolley KA. 2015. PubMLST.org--The new home for the Borrelia MLSA database. Ticks Tick Borne Dis. 6(6):869-871
- Becker NS, Margos G, Blum H, Krebs S, Graf A, Lane RS, Castillo-Ramírez S, Sing A, Fingerle V. 2016 Recurrent evolution of host and vector association in bacteria of the Borrelia burgdorferi sensu lato species complex. BMC Genomics 17(1):734
- Meinel DM, Margos G, Konrad R, Krebs S, Blum H, Sing A. 2014. Next generation sequencing analysis of nine Corynebacterium ulcerans isolates reveals zoonotic transmission and a novel putative diphtheria toxin-encoding pathogenicity island. Genome Med.;6(11):113
- Meinel DM, Kuehl R, Zbinden R, Boskova V, Garzoni C, Fadini D, Dolina M, Blümel B, Weibel T, Tschudin-Sutter S, Widmer AF, Bielicki JA, Dierig A, Heininger U, Konrad R, Berger A, Hinic V, Goldenberger D, Blaich A, Stadler T, Battegay M, Sing A, Egli A. 2016. Outbreak investigation for toxigenic Corynebacterium diphtheriae wound infections in refugees from Northeast Africa and Syria in Switzerland and Germany by whole genome sequencing. Clin Microbiol Infect. 22(12):1003.e1-1003.e8
- Margos G, Hepner S, Mang C, Marosevic D, Reynolds SE, Krebs S, Sing A, Derdakova M, Reiter MA, Fingerle V. 2017. Lost in plasmids: next generation sequencing and the complex genome of the tick-borne pathogen Borrelia burgdorferi. BMC Genomics 30, 18(1):422
- Margos G, Hepner S, Mang C, Sing A, Liebl B, Fingerle V. 2017. Completed Genome Sequences of Borrelia burgdorferi Sensu Stricto B31(
PHM) and Closely Related Patient Isolates from Europe. Genome Announc 13, 5(28) - Marosevic D, Margos G, Wallich R, Wieser A, Sing A, Fingerle V. 2017. First insights in the variability of Borrelia recurrentis genomes. PLoS Neglect Trop Dis 11(9):e0005865.
- Meinel DM, Heinzinger S, Eberle U, Ackermann N, Schönberger K, Sing A. 2018 Whole genome sequencing identifies influenza A H3N2 transmission and offers superior resolution to classical typing methods. Infection 46(1):69-76
- Dangel A, Berger A, Konrad R, Bischoff H, Sing A. 2018. Next Generation Sequencing outbreak investigation of geographical clusters of non-toxigenic Corynebacterium diphtheriae cases in Germany. Emerg Infect Dis (im Druck)