
- Startseite >>
- Tiergesundheit >>
- Tierkrankheiten >>
- Tierkrankheiten A Z >>
- Bovine Spongiforme Enzephalopathie
Bovine spongiforme Enzephalopathie (BSE)
Erreger
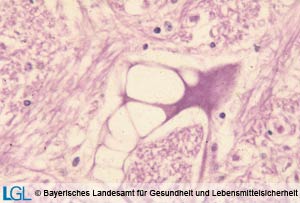
Abbildung 1: Mikroskopische Aufnahme eines Rindergehirnes mit Nachweis der typischen Vakuolen in Nervenzellen bei Vorliegen von Boviner spongiformer Enzephalopathie.
BSE gehört zur Gruppe der übertragbaren spongiformen Enzephalopathien (TSE - Transmissible spongiforme Enzephalopathie) die durch ein fehlgefaltetes Protein, ein sog. pathologisches Prion ausgelöst wird. BSE ist eine infektiöse neurodegenerative Prionenerkrankung des Rindes, die erstmalig 1986 in Großbritannien nachgewiesen wurde. Scrapie bei Schaf und Ziege ist ebenfalls eine analoge Prionerkrankung und wurde bereits im 18. Jahrhundert in England beschrieben. Diese Prionenerkrankungen werden durch infektiöse Proteine verursacht, die ihre fehlgefaltete, pathologische Konformation auf andere Proteine übertragen können. Im Gegensatz zu allen anderen Krankheitserregern wie Viren, Bakterien oder Pilzen sind Prionen keine Lebewesen und enthalten daher auch keine Nukleinsäure (Erbinformation).
Grundsätzlich kann man zwei Formen des Prion-Proteins unterscheiden: eine harmlose, zelluläre Form des Prion-Proteins (PrPC für c=cellular), die jeder Mensch und jedes Säugetier auf fast jeder Zelle trägt, und deren infektiöse Isoform PrPSc (Sc=Scrapie) oder PrPBSE genannt. Die einzelnen Aminosäurebausteine, aus denen die beiden Formen bestehen, sind identisch. Die beiden Formen unterscheiden sich lediglich aufgrund ihrer räumlichen Struktur. Während bei der zellulären Form α-Helix-Spiralen dominieren, faltet sich die pathologische Form vor allem zu ß-Faltblattstrukturen. Im Ablauf des Krankheitsgeschehens kommt es zur Umfaltung: α-Helix-Spiralen gehen verloren und ß-Faltblattstrukturen dominieren. Aus dem harmlosen physiologischen Prion-Protein entsteht die fehlgefaltete pathologische Isoform.
Vorkommen und Übertragung
BSE wurde im Vereinigten Königreich erstmals 1986 nachgewiesen und die BSE-Fälle erreichten dort bereits in den Jahren 1993/94 ihren Höhepunkt , während in den anderen EU-Mitgliedsstaaten die meisten Fälle 2001/02 aufgetreten sind. Die Verfütterung von unzureichend erhitztem Risikomaterial - Gehirn und Nervengewebe - wurde bald als Quelle der BSE-Erkrankungen festgestellt und in der Folge durch entsprechende gesetzliche Regelungen verboten. In Deutschland wurde 2001 mit 125 Fällen der Höhepunkt der nachgewiesenen BSE-Fälle erreicht. In den Jahren 2008 und 2009 wurden jeweils nur noch zwei Fälle von übertragbarer BSE bei Rindern diagnostiziert.
Atypische BSE tritt in seltenen Fällen spontan bei älteren Tieren auf, wird also nicht wie die klassische BSE durch die Verfütterung unzureichend erhitzter Wiederkäuerfette und -proteine, die das krankmachende Prion-Protein enthielten, an Rinder verursacht. Die Verteilung der Fälle und das Auftreten bei älteren Tieren sprechen für eine spontane Entstehung, so dass immer wieder mit einem Fall gerechnet werden muss. Die letzten beiden Fälle in Deutschland sind 2014 in Brandenburg aufgetreten, aktuell (2021) gibt es einen Fall in Bayern. Am Tierseuchenstatus ändert sich aufgrund dieses Falles in Deutschland nichts. Europaweit traten ca. 97% aller diagnostizierten BSE-Fälle im Vereinigten Königreich auf.
BSE ist auf verschiedene Wirte (Versuchstiere, Wiederkäuer, Fleischfresser, Affen) übertragbar, u. a. auf den Menschen (Zoonose). Besonders gefürchtet ist die Übertragung des BSE-Prion, das bei einigen jungen Menschen erstmals 1996 in England nach relativ kurzen Inkubationszeiten zur Krankheit mit Todesfolge geführt hat und als variante Form der Creutzfeldt-Jakob Krankheit (vCJD) bezeichnet wird. Ansonsten hat die BSE wie andere TSE-Erkrankungen eine sehr lange Inkubationszeit.
Krankheitsbild
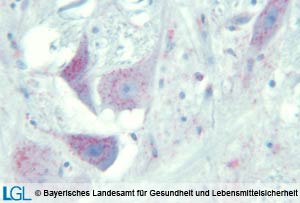
Abbildung 2: Mikroskopische Aufnahme des Gehirnes eines Geparden mit immunhistologischem Nachweis von Prion-Protein
BSE führt zu fortschreitenden krankhaften Veränderungen des zentralen Nervensystems. Nach einer langen Inkubationszeit werden zentralnervöse Symptome wie Koordinationsstörungen und Demenz beobachtet, in deren Folge immer der Tod eintritt.
Das mikroskopische Bild des Gehirns ergibt ein weitgehend einheitliches Bild, das durch den Untergang einer Zellpopulation und durch die Akkumulation von Protein gekennzeichnet ist.
Als ursächliches Agens vermutet man das sogenannte infektiöse Prion-Protein. Der endgültige Beweis für diese Theorie ist jedoch noch nicht geführt. TSE findet man sowohl beim Tier als auch beim Menschen. In Abhängigkeit von der Wirtsspezies und der Pathologie werden die TSE-Erkrankungen unterschiedlich bezeichnet.
Die pathologischen Erscheinungen der BSE sind charakterisiert als neuro-degenerative Veränderungen im Zentralnervensystem, die Nervenzellzerstörung zeigt im histologischen Bild ein schwammartiges (spongiformes) Aussehen. Die Erkrankung verläuft immer tödlich und ist z. Z. nicht therapierbar.
TSE bei Tieren und beim Menschen
Beim Tier kennt man fünf verschiedene Formen der TSE-Erkrankung: Am längsten bekannt ist die Krankheit „Scrapie“, auch Traberkrankheit der Schafe und Ziegen, die den Prototyp der TSE-Erkrankungen darstellt. Daneben sind die Transmissible Mink Enzephalopathie der Nerze (TME), die Chronic Wasting Disease der Hirsche (CWD), die Bovine Spongiforme Enzephalopathie (BSE) und die Feline Spongiforme Enzephalopathie (FSE) bekannt.
Scrapie wurde erstmals 1732 bei Schafen beschrieben. Das klinische Bild ist im typischen Fall gekennzeichnet durch starken Juckreiz (engl.: to scrape, kratzen), Ataxien (Traberkrankheit), Tremor (franz.: la tremblante, Zittern) und Übererregbarkeit. Im Endstadium tritt der völlige Koordinationsverlust ein und die Tiere liegen fest. Eine Gefährdung des Menschen durch Scrapie gilt aufgrund intensiver Forschungsarbeiten nach heutigem Kenntnisstand als ausgeschlossen.
BSE wurde erstmals 1986 in Großbritannien (GB) nachgewiesen. Als Infektionsursache wurde die Verfütterung von unzureichend erhitztem und mit Prionen kontaminiertem Tiermehl an Rinder vermutet. Daraufhin wurde 1988 die Verfütterung von wiederkäuerhaltigem Tiermehl an Wiederkäuer verboten. In der EU wurde im Jahr 1994 ein Tiermehl-Verfütterungsverbot für Wiederkäuer eingeführt und 2001/2002 zum totalen Tiermehl-Verfütterungsverbot für weitere Tierarten ausgeweitet. Übertragungsversuche an Rindern ergaben, dass die Infektiosität in erster Linie auf das ZNS, die Netzhaut und den hinteren Dünndarmabschnitt beschränkt ist. Diese sog. „spezifizierten Risikomaterialien (SRM)“ werden deshalb aus Gründen des vorbeugenden gesundheitlichen Verbraucherschutzes im Rahmen der Schlachtung altersabhängig entfernt und unschädlich beseitigt.
Von der oben beschriebenen klassischen BSE wird die atypische Form der BSE aufgrund biologischer Erregereigenschaften unterschieden. Da sich die Krankheitssymptome beider BSE-Formen gleichen, kann die Unterscheidung nur mittels labordiagnostischer Methoden erfolgen.
Die beim Menschen bereits lange bekannten TSE-Erkrankungen werden aufgrund ihrer Genese in zwei Rubriken gegliedert. In der Gruppe der erblich bedingten TSE-Erkrankungen werden die Fatale Familiäre Insomnie (FFI), das Gerstmann-Sträussler-Scheinker-Syndrom (GSS) und die sog. klassische Form der Creutzfeldt-Jakob-Krankheit (CJK) unterschieden. Daneben bilden die sog. neue Variante der Creutzfeldt-Jakob-Krankheit (nVCJK) und die „Kuru“ Krankheitsbilder mit rein übertragbaren Ursachen die zweite Gruppe.
Während die klassische CJK vor allem ältere Patienten betrifft, tritt die nVCJK bislang nur bei jüngeren Patienten auf und ist vermutlich im Zusammenhang mit auf den Verzehr von mit Prionen kontaminierter Nahrung zurückzuführen. Bislang ist ein Ausbruch der nVCJK auf Menschen beschränkt, deren Prion-Gen einen bestimmten Genotyp aufweist..
Diagnostik und Prophylaxe
Für die postmortale Diagnosestellung ist die Histopathologie ein sicherer Nachweis. An Gehirnmaterial (Kleinhirnstamm) von Schlachttieren werden ELISA-Schnelltest durchgeführt, als Bestätigungstestdient der Western Blot (Immunblot).
Am lebenden Tier ist die Diagnose BSE schwierig zu stellen, weil andere zentralnervöse Erkrankungen ähnliche Symptome auslösen können. Verschiedene Systeme zur klinischen BSE-Diagnostik am lebenden Rind wurden erprobt.
Ein BSE-Verdacht wird aufgrund klinischer Symptomatik, einer histopathologischen Untersuchung oder eines wiederholt reaktiven Ergebnisses in einem der national und von der EU zugelassenen BSE-Schnelltests geäußert. BSE-Verdachtsfälle müssen am Nationalen Referenzlabor (NRL) für BSE/Scrapie am FLI mittels der von der EU zugelassenen Methoden bestätigt werden.
Die seit 2001 ergriffenen Maßnahmen zur Bekämpfung dieser Erkrankung (Verbot der Verfütterung tierischen Eiweißes an Säugetiere, Definition und Entfernung spezifizierter Risikomaterialien, großflächige BSE-Schnelltestuntersuchungen) haben sich als wirksam erwiesen.
Das Nationale Referenzlabor für TSE: Friedrich-Loeffler-Institut, 17493 Greifswald - Insel Riems (Nationales Referenzlabor für neue und neuartige Tierseuchenerreger, Tel. 038351 7-1165).
Gesetzliche Grundlagen
- Verordnung (EG) 999/2001
- TSE-Überwachungsverordnung