
- Startseite >>
- Tiergesundheit >>
- Tierkrankheiten >>
- Tierkrankheiten A Z >>
- Bovine Spongiforme Enzephalopathie
Hintergrundinformation: BSE/TSE
Was ist Bovine Spongiforme Enzephalopathie / TSE?
Die BSE (Bovine Spongiforme Enzephalopathie) gehört zur Gruppe der übertragbaren, schwammartigen, nicht entzündlichen Gehirnerkrankungen (Transmissible Spongiforme Enzephalopathien, TSE), die zu fortschreitenden krankhaften Veränderungen des zentralen Nervensystems führen. Nach einer langen Inkubationszeit werden zentralnervöse Symptome wie Koordinationsstörungen und Demenz beobachtet, in deren Folge immer der Tod eintritt. Das mikroskopische Bild des Gehirns ergibt ein weitgehend einheitliches Bild, das durch den Untergang einer Zellpopulation und durch die Akkumulation von Protein gekennzeichnet ist. Sowohl klassische Erreger wie Bakterien oder Viren, als auch nackte Erbsubstanz werden nach dem derzeitigen Wissensstand als Krankheitserreger ausgeschlossen. Als ursächliches Agens vermutet man das sogenannte infektiöse Prion-Protein (proteinacous infectious particle), welches die fehlgefaltete Isoform eines natürlich vorkommenden Proteins darstellt und nicht der klassischen Definition eines Infektionserregers entspricht. Der endgültige Beweis für diese Theorie ist jedoch noch nicht geführt. TSE-Erkrankungen findet man sowohl beim Tier als auch beim Menschen. In Abhängigkeit von der Wirtsspezies und der Pathologie werden die TSE-Erkrankungen unterschiedlich bezeichnet.
TSE beim Tier
Beim Tier kennt man fünf verschiedene Formen der TSE-Erkrankung: Am längsten bekannt ist die Krankheit „Scrapie“, auch Traberkrankheit der Schafe und Ziegen, die den Prototyp der TSE-Erkrankungen darstellt. Daneben sind die Transmissible Mink Enzephalopathie der Nerze (TME), die Chronic Wasting Disease der Hirsche (CWD), die Bovine Spongiforme Enzephalopathie (BSE) und die Feline Spongiforme Enzephalopathie (FSE) zu berücksichtigen. Außerdem wurden TSE-Erkrankungen bei in zoologischen Gärten lebenden Wildwiederkäuern und Großkatzen beschrieben.
Scrapie wurde erstmals 1732 bei Schafen beschrieben. Das klinische Bild ist im typischen Fall gekennzeichnet durch starken Juckreiz (engl.: to scrape, kratzen), Ataxien (Traberkrankheit), Tremor (franz.: la tremblante, Zittern) und Übererregbarkeit. Im Endstadium tritt der völlige Koordinationsverlust ein und die Tiere liegen fest. Es werden auch klinische Fälle beschrieben, bei denen der Juckreiz fehlt und Apathie (Teilnahmslosigkeit) im Vordergrund steht. Seit den 30er Jahren weiß man, dass die Erkrankung sich experimentell auf Schafe und Ziegen übertragen lässt. Die natürlichen Übertragungswege der Scrapie sind bisher noch nicht vollständig geklärt. Erregerhaltige Nachgeburt wird heute als Infektionsquelle für die horizontale Ausbreitung innerhalb einer Herde angesehen. Isländische Studien belegen, dass kontaminierte Böden als Infektionsquellen berücksichtigt werden müssen, da der Erreger darin länger als drei Jahre stabil bleibt. Die Inkubationszeit beträgt mindestens ein Jahr und variiert in Abhängigkeit vom Genotyp des Empfängertieres. Die klinische Phase dauert bei Schafen vier bis neun Monate, bei Ziegen ist sie häufig kürzer. Eine Gefährdung des Menschen durch Scrapie gilt aufgrund intensiver Forschungsarbeiten nach heutigem Kenntnisstand als ausgeschlossen. Scrapie kommt weltweit vor. Lediglich Australien und Neuseeland gelten als scrapiefrei. In Bayern treten erfahrungsgemäß wenige Einzelfälle pro Jahr auf. So wurden im Verlauf der letzten vierzehn Jahre jeweils acht Fälle (2006), drei Fälle (2007), ein Fall (2008), kein Fall (2009), zwei Fälle (2010 und 2011), ein Fall (jeweils 2012 und 2013), zwei Fälle (jeweils 2014 und 2015),ein Fall (2016), kein Fall (2017, 2018 und 2019) nachgewiesen. Im Jahr 2020 trat bislang im Februar ein Fall auf (Quelle: http://www.bmel.de/DE/Tier/Tiergesundheit/Tierseuchen/_texte/TSE-FaelleGesamt.html).
Die TME der Nerze wurde erstmals 1947 auf einer Nerzfarm in Wisconsin beobachtet. Seither wurden vereinzelt Ausbrüche in Kanada, Finnland, Russland und der ehemaligen DDR gemeldet. Als Ursache der TME wird die Verfütterung von kontaminiertem Tiermehl vermutet.
Die CWD bei Hirschartigen wurde erstmals 1967 bei in Gefangenschaft gehaltenen nordamerikanischen Maultierhirschen und Wapiti beschrieben, kommt jedoch auch in der freien Wildbahn vor. In der Natur ist der Ursprung der Erkrankung noch ungeklärt. Die Übertragung erfolgt unter den Wildtieren horizontal, vermutlich über Kot und Speichel.
Die BSE wurde erstmals 1986 in Großbritannien (GB) beschrieben. Als Infektionsursache wurde die Verfütterung von unzureichend erhitztem und mit Prionen kontaminiertem Tiermehl an Rinder vermutet. Daraufhin wurde 1988 die Verfütterung von wiederkäuerhaltigem Tiermehl an Wiederkäuer verboten. In der EU wurde im Jahr 1994 ein Tiermehl-Verfütterungsverbot für Wiederkäuer eingeführt und 2001/2002 zum totalen Tiermehl-Verfütterungsverbot für weitere Tierarten ausgeweitet. Das Auftreten einzelner BSE-Fälle bei Jungtieren, die in GB nach dem totalen Verfütterungsverbot geboren wurden ließ zunächst auch eine Übertragung an Nachkommen infizierter Muttertiere vermuten. Experimentell konnte der Nachweis für eine vertikale Übertragung bislang aber nicht erbracht werden. Übertragungsversuche an Rindern ergaben, dass die Infektiosität in erster Linie auf das ZNS, die Netzhaut und den hinteren Dünndarmabschnitt beschränkt ist. Diese sog. „spezifizierten Risikomaterialien (SRM)“ werden deshalb aus Gründen des vorbeugenden gesundheitlichen Verbraucherschutzes im Rahmen der Schlachtung altersabhängig entfernt und unschädlich beseitigt. Die Wirkung dieser Maßnahmen spiegelt sich seither in einem starken und kontinuierlichen Rückgang der Neuerkrankungen wieder. Nach dem Höhepunkt der Epidemie im Jahr 1992 mit 37.280 registrierten Fällen in GB, sank die Anzahl der Neuerkrankungen stetig. 2003 wurden in GB noch 611 Fälle registriert, seit 2011 treten im Jahresdurchschnitt weniger als 10 Fälle auf (Quelle: http://www.oie.int/en/animal-health-in-the-world/bse-specific-data/). BSE kommt heute weltweit nur noch selten vor (<20 registrierte Fälle pro Jahr).
Von der oben beschriebenen klassischen BSE wird die atypische Form der BSE aufgrund biologischer Erregereigenschaften unterschieden. Da sich die Krankheitssymptome beider BSE-Formen gleichen, kann die Unterscheidung nur mittels labordiagnostischer Methoden erfolgen. Aufgrund der unterschiedlichen Masse der Prion-Proteine wird die atypische Form der BES in die zwei Typen „H“ (high) und „L“ (low) unterteilt. Die atypische BSE wurde bisher in Kanada, USA, Japan und in der EU registriert. Da die Fälle bislang sehr selten und nur bei Tieren ab einem Alter von acht Jahren auftreten, wird eine spontane Entstehung angenommen. Daher ist auch zukünftig mit dem Auftreten von Einzeltiererkrankungen zu rechnen. In Deutschland traten zuletzt im Januar und Februar 2014 zwei atypische Fälle von BSE auf. Am 09.01.2014 wurde bei einem zehn Jahre alten Schlachtrind die atypische BSE, Typ-L, und am 05.02.2014 bei einem 11 Jahre alten Schlachtrind der Typ-H festgestellt. Zuvor war bei zwei deutschen Schlachtrindern in den Jahren 2002 und 2004 die atypische BSE diagnostiziert worden. Die Rinder waren zum Schlachtzeitpunkt 15 und 13 Jahre alt (Quelle: http://www.fli.bund.de/fileadmin/dam_uploads/Publikationen/FLI-Informationen/FLI-Information_Atypische_BSE_20120307.pdf).
Im Zuge der BSE-Bekämpfung wurden 1990 erstmals auch Fälle von TSE bei Hauskatzen beobachtet (FSE). Daneben erkrankten Pumas, Ozelote und Tiger in britischen Zoos. Als Krankheitsursache wird die Verfütterung von mit Prionen kontaminiertem Fleisch und Tiermehl angenommen.
TSE beim Menschen
Die beim Menschen bereits lange bekannten TSE-Erkrankungen werden aufgrund ihrer Genese in zwei Rubriken gegliedert. In der Gruppe der erblich bedingten TSE-Erkrankungen werden die Fatale Familiäre Insomnie (FFI), das Gerstmann-Sträussler-Scheinker-Syndrom (GSS) und die sog. klassische Form der Creutzfeldt-Jakob-Krankheit (CJK) unterschieden. Daneben bilden die sog. neue Variante der Creutzfeldt-Jakob-Krankheit (nVCJK) und die „Kuru“ Krankheitsbilder mit rein übertragbaren Ursachen.
Die klassische Form der CJK kommt mit einer Häufigkeit von 1 Fall pro 1 Mio. Einwohner und Jahr vor. Zu 80% tritt die sog. „sporadische Form“ auf, deren Ursache nicht ermittelt werden kann. Im Weiteren gibt es Erkrankungen, die mit Veränderungen im Gen des körpereigenen Prionproteins erklärt werden können. Auch iatrogene Fälle wurden beschrieben, die nach Organtransplantationen von CJK-positiven Spendern oder durch unzureichende Sterilisation neurochirurgischer Instrumente auftraten. Während die klassische CJK vor allem ältere Patienten betrifft, tritt die nVCJK bislang nur bei jüngeren Patienten auf und ist vermutlich im Zusammenhang mit BSE auf den Verzehr von mit Prionen kontaminierter Nahrung zurückzuführen. Bislang ist ein Ausbruch der nVCJK auf Menschen beschränkt, deren Prion-Gen einen bestimmten Genotyp aufweist.
Die Kuru, Lach- oder Schüttelkrankheit genannt, wurde erstmals 1957 bei einem Eingeborenenstamm in Papua Neuguinea beschrieben. Dort wurden Frauen und Kinder in einem kannibalischen Ritual mit dem Gehirn von Toten eingerieben. Diese infizierten sich durch die Aufnahme von Prionen über die Schleimhäute. Daneben entstanden Erkrankungen durch Endokannibalismus. Seit dem Verbot des Kannibalismus wurden keine Neuerkrankungen mehr registriert.
Das Prion-Protein
Grundsätzlich kann man zwei Formen des Prionproteins unterscheiden: eine harmlose, zelluläre Form des Prionproteins (PrPC für c=cellular), welches jeder Mensch und jedes Säugetier auf fast jeder Zelle trägt, und deren infektiöse Isoform PrPSc (Sc=Scrapie) oder PrPBSE genannt. Die einzelnen Aminosäurebausteine, aus denen die beiden Formen bestehen, sind identisch. Die beiden Formen unterscheiden sich lediglich aufgrund ihrer räumlichen Struktur. Während bei der zellulären Form a-Helix-Spiralen dominieren, faltet sich die pathologische Form vor allem zu ß-Faltblattstrukturen. Im Ablauf des Krankheitsgeschehens kommt es zur Umfaltung: a-Helix-Spiralen gehen verloren und ß-Faltblattstrukturen dominieren. Aus dem harmlosen physiologischen Prionprotein entsteht die fehlgefaltete pathologische Isoform.
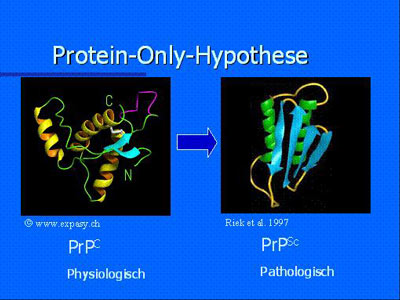
Nachweis des pathologischen Prionproteins
Zum Nachweis machen sich die momentan in Deutschland zugelassenen Schnelltests die unterschiedlichen physikochemischen Eigenschaften der beiden Proteine zunutze.
Während PrPC durch das Enzym Proteinase K vollständig verdaut wird, wird PrPBSE bzw. PrPSc nur unvollständig verdaut, ein Rest bleibt nachweisbar. Ein anderes Schnelltestverfahren nutzt das unterschiedliche Bindungsverhalten von PrPBSE.